Adsorption Locator-定位SO2分子在Ni(111)表面的吸附位置
目的:介绍利用Adsorption Locator模块研究不同类型吸附质在不同性质衬底上吸附的计算方法。
所用模块:Materials Visualizer、Adsorption Locator、Forcite
先决条件:需要先掌握利用Materials Visualizer绘制苯甲酰胺分子的教程研究背景:此教程基于Harrison等人(2006年)发表的一篇论文“Cu(111)和Ni(111)表面SO2和SO3结构的密度泛函理论研究”(Density functional theory investigation of the structure of SO2 and SO3 on Cu(111) and Ni (111))。研究SO2和SO3分子的吸附,是基于电站排放物中清除此类环境污染物的需求。本文对SO2分子吸附的几种几何构型进行了DFT计算。可以通过以下两种方式将Adsorption Locator模块作为初始构型准备和筛选工具:
自动生成吸附构型,然后可以将其用作进一步DFT计算的初始构型,如上述论文中所述;
使用力场方法获得每个生成构型的能量排序,并得到最佳吸附位置。
本教程针对Ni(111)上的SO2的情况进行计算,包含如下几部分:开始、构建结构模型、设置Adsorption Locator计算、结果分析、结论。
注意:为了确保您可以完全按照预期的方式学习本教程,您应该使用“设置管理器(Settings Organizer)”对话框确保项目中所有参数都设置为BIOVIA的默认值。
1、开始
首先启动Materials Studio并创建一个新项目。
打开“新建项目New Project”对话框,输入Ni_SO2作为项目名称,单击“确定OK”按钮。于此创建一个新项目,且Ni_SO2在项目浏览区域Project Explorer中列出。
2、构建结构
在本节教程中,将构建Ni(111)表面和SO2吸附质的结构。
(1)构建SO2分子结构。
选择File | New...从菜单栏打开新建文档New Document对话框,双击3D Atomistic。使用绘制工具,构建一个SO2分子,将两个S-O键定义为双键,单击“清除Clean按钮。从菜单栏中选择File | Save并将结构保存为SO2.xsd。
(2)在将SO2分子吸附到将要构建的Ni表面之前,对其进行优化。
从菜单栏中选择Modules | Forcite | Calculation,打开Forcite Calculation对话框。在Setup选项卡上,将任务task更改为几何优化Geometry Optimization,将精度Quality更改为高Fine。选择Energy选项卡并将力场Forcefield更改为COMPASSIII。单击运行Run按钮。
在计算任务完成后,将在SO2 Forcite GeomOpt/SO2.xsd中创建优化后的结构。
(3)构建Ni(111)表面。从菜单栏中选择File | Import...打开导入文档Import Document对话框。导航到Structures/metals/pure metals文件夹,并导入Ni.xsd模型。
从菜单栏中选择Build | Surfaces | Cleave Surface,打开Cleave Surface对话框。将Cleave plane (h k l)更改为1 1 1,并按下TAB键。将Fractional Thickness增加到6.0,然后按TAB键。
单击Cleave按钮并关闭对话框。表面结构中的层数选择,要使表面厚度大于计算中使用的非键项截断值(截断值的选择需要权衡精度和计算所需时间),这里使用6层Ni原子层,可以使SO2分子只与表面层中的Ni原子发生非键相互作用,而不会不合理地增加计算时间。
(4)构造一个2×2的超晶胞,使得表面更加接近真实的表面,用于吸附SO2分子。从菜单栏中选择Build | Symmetry | Supercell,打开Supercell对话框。将U和V的值增加到2,然后单击Create Supercell按钮,关闭对话框。
(5)将此结构转换为具有三维周期性的结构。由于计算采用三维周期性边界条件,真空层的大小要足够大,以使吸附质的非键项计算不会与表面底层原子的周期图像相互作用。真空层厚度的选取需要基于非键截断和吸附质分子构型,并适当进行提高,以确保吸附质只与表面相互作用。
在本教程的情况下,Ni表面层和真空以外的下一层之间15 Å的距离是足够的。这防止了下一个重复单元和Ni表面或SO2分子之间的任何非键相互作用。
为了更清楚地显示Ni原子,可使用球棍模型显示。
从菜单栏中选择View | Display style打开Display Style对话框。在Atom选项中,将显示样式Display style更改为Ball and stick,然后关闭对话框。
3、设置Adsorption Locator模块计算参数
(1)现在可以开始Adsorption Locator模块的计算,设置的参数主要包括“精度”(quality)和“力场”(forcefield),以及选择要在计算中检测吸附位置的方法。
单击“模块Modules”工具栏上的Adsorption Locator模块后的箭头,然后从下拉列表中选择计算Calculation以打开Adsorption Locator Calculation对话框,该过程中要确保Ni (1 1 1).xsd是活动文档,处于打开状态。
在Setup选项卡上,设置精度Quality为Fine。从吸附质Adsorbate下拉列表中选择SO2 Forcite GeomOpt/SO2.xsd,将加载数量Loading设置为1。
在Energy选项卡上,从“力场Forcefield”下拉列表中选择COMPASSIII。
在Location选项卡上,选中“由原子集合定义的表面区域Surface region defined by atom set”复选框。
当计算精度Quality设为Fine时,计算中将进行多次模拟退火循环,每个循环运行多步,以获得更精确的统计信息。(2)接下来选择原子,从而定义表面区域。
选择顶层的Ni原子,被选中的原子显示为黄色。
在Adsorption Locator Calculation对话框的Location选项卡中,单击Add按钮,从而将选定的原子包含在为吸附计算设置的目标原子集合TargetAtoms set中。选中“设置最大吸附距离Set maximum adsorption distance”复选框,输入设定值5.0。
该方法对从选定目标原子到吸附质包裹体指定的最大距离定义的区域进行取样。设定成5.0允许体系创建吸附在Ni表面的SO2的新构型。如果最大吸附距离太短,则无法创建某些构型,如果太长,则不会添加新构型。
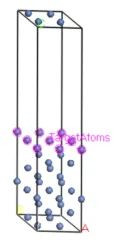
定义了目标原子集合后的Ni(111)表面
(3)固定能量窗口(fixed energy window)用于选择报告哪些构型。在这种情况下,这将返回与最低构型能量相差小于100 kcal/mol的所有构型。在初始计算中,可以使用更宽的能量窗口,因为这样可以确保不会遗漏任何重要构型。
在Properties选项卡上,选中所有属性all properties复选框。选择Fixed energy window选项,并输入100 kcal/mol的值。
(4)现在可以运行计算了。
单击Run按钮并关闭对话框。
4、结果分析
计算开始后,Materials Studio将创建一个名为Ni (1 1 1) Adsorption Anneal的新文件夹。计算完成后,所有结果将会返回到此文件夹中。
打开3D原子文档Ni (1 1 1) Fields.xsd。
这个文件显示了吸附点的区域,密度越高的点是越可能的吸附位置。
在文档中单击鼠标右键,然后从快捷菜单中选择“显示样式Display Style”以打开Display Style对话框。在Field选项卡上,选择“按域值显示颜色Color by field values”选项和体显示样式Volume 显示样式,关闭对话框。
这将更可能的吸附位置显示为绿色,而不太可能的吸附位置显示为红色。
打开表格文件Ni (1 1 1).std。
表格文件中包含了最低能量构型的结构和能量。可以将本教程得到的结果与参考文献中得到的最低值进行比较,也可用Cu代替Ni重复一遍Adsorption Locator计算。5、总结Adsorption Locator模块的计算结果与以下类似。
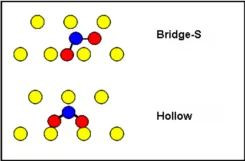
两个可能的结合位置示意图
提示:为更明显显示Ni表面能量场的视图,请选择所有非表面Ni原子,并将其显示样式设置为“无None”。因为蒙特卡罗方法有一个统计分量,得到的结果可能会略有不同。要获得重复性较高的结果,可运行时间更长、精度更高的模拟。Ni上吸附的SO2位于桥位的能量最低,这与Harrison等人的论文一致。该论文还报道了SO2在Cu表面的桥位和空位的吸附,两者之间的能量没有明显的差异。本教程到此结束。
参考文献:"Density functional theory investigation of the structure of SO2 and SO3 on Cu(111) and Ni(111)" by M. J. Harrison, D. P. Woodruff, and J. Robinson, Surface Science, 600, 1827, (2006).